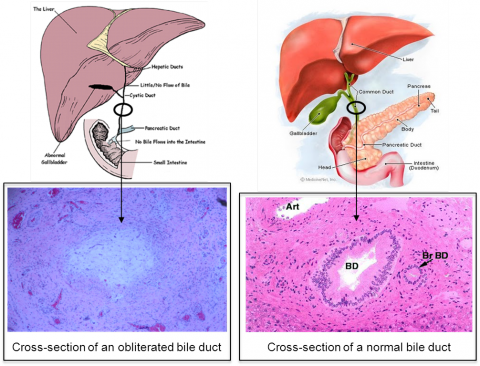
 Biliary atresia is a rare newborn liver disease resulting from a rapid and progressive obliteration of large bile ducts- the tubes draining bile from the liver to the intestine- that begins in the perinatal period and become completely obstructed by four months of age (see image below).
Biliary atresia is a rare newborn liver disease resulting from a rapid and progressive obliteration of large bile ducts- the tubes draining bile from the liver to the intestine- that begins in the perinatal period and become completely obstructed by four months of age (see image below).
In Canada, biliary atresia occurs in 1:19,000 live births, accounting for 18-22 new cases each year in our country. While not an inherited disease, the precise factors that cause biliary atresia remain poorly understood. Yet the condition uniquely manifests in newborns by three weeks of age (not ever in older infants, children or adults), with persistent jaundice in association with pale (acholic) stools.
The current standard of care for affected infants is sequential surgical treatment with an initial Kasai procedure (KP) at the time of diagnosis, followed by liver transplantation for those patients who progress to cirrhosis. Left untreated, 100% of BA cases will be dead by age three. At the KP, the obstructed bile duct is removed and a loop of bowel is brought to the porta of the liver to serve as a new conduit for bile drainage. A successful KP results in the clearance of jaundice and the return of pigment to the stool. If the KP is unsuccessful or not performed because of very late presentation, liver transplantation is necessary for survival.
Several outcome studies have demonstrated that the patient age at the time of the KP is a key prognostic factor. The older the infant at the time of KP, the less favourable is the long-term survival with their own liver. In Canada, if the KP is performed at <30 days of age, the 10 yr patient survival with their own liver is 50%. In contrast, the patient survival with their own liver is <20% for those cases having the KP at > 90 days of age. Yet, only 8% of affected Canadian infants have the KP at <30 days of age with almost 20% presenting late having the KP at >90 days of age. Timely diagnosis and early KP for BA remains a challenge in Canada. There are several reasons: jaundice in BA is often mistakenly considered inconsequential and no further investigations are completed, the assessment of stool colour is not standard practice for health care providers or parents, and there are currently no screening tests for the diagnosis of BA.
Evidenced based standards for best management practices are not yet available. The goal of CBAR is to optimize care and improve survival rates for children with biliary atresia.


